Delgado-Marín M, Sánchez S, Cook A, Castro M, López J, Hernández I, Zamorano JL, Zaragoza C, Saura M. ILK-Dependent Modulation of DPP4 Prevents Progression of Calcific Aortic Valve Disease
Arterioscler Thromb Vasc Biol. 2026
"By linking endothelial ILK loss to DPP4-driven inflammation and calcific remodeling, this study identifies a clinically actionable pathway in the early progression of calcific aortic valve disease". - Dr María Delgado-Marín & Dr Marta Saura
Summary:
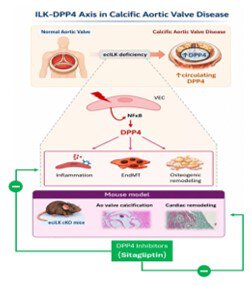
Background: Calcific aortic valve disease (CAVD) is characterized by endothelial dysfunction, fibrosis, and osteogenic calcification, yet the molecular mechanisms driving disease progression remain incompletely understood, and no pharmacological therapies are currently available. Reduced endothelial ILK (integrin-linked kinase) expression has been implicated in CAVD, but its downstream effectors remain undefined.
Methods: To explore the direct role of ILK in CAVD development, we used a mouse model in which ILK is conditionally deleted from endothelial cells (endothelial cell-specific ILK conditional knockout). DPP4 (dipeptidyl peptidase 4) expression and activity were examined in human aortic valve tissue and plasma from patients with and without CAVD as a potential therapeutic target. Mechanistic studies were performed in human valvular endothelial cells subjected to ILK silencing, with or without pharmacological or genetic DPP4 inhibition. In vivo, endothelial cell-specific ILK conditional knockout mice were treated with the DPP4 inhibitor sitagliptin, and valvular, cardiac function, and remodeling were assessed.
Results: DPP4 expression was increased in circulation and in the aortic valves of patients with CAVD and was inversely correlated with ILK levels, with predominant localization to valvular endothelial cells. ILK silencing in human valvular endothelial cells increased DPP4 expression and enzymatic activity, promoted endothelial-to-mesenchymal transition, and induced osteogenic reprogramming; these effects were attenuated by DPP4 inhibition. Endothelial ILK deletion in mice recapitulated CAVD features, including inflammation, valve thickening, calcification, and cardiac remodeling, all of which were associated with increased DPP4. Sitagliptin treatment mitigated disease severity. Mechanistically, sitagliptin inhibited NF-κB (nuclear factor kappa B)-driven DPP4 upregulation caused by ILK loss.
Conclusions: These findings identify DPP4 as a key downstream effector linking endothelial ILK deficiency to inflammation, endothelial-to-mesenchymal transition, and calcific remodeling in CAVD and support DPP4 inhibition as a potential disease-modifying strategy to slow disease progression.
Why do you highligth this publication?
We highlight this publication because it identifies the DPP4/ILK axis as a key mechanistic link between endothelial dysfunction and calcific aortic valve disease (CAVD), a highly prevalent condition for which no pharmacological treatment is currently available.
This study shows that endothelial ILK deficiency promotes DPP4 upregulation, inflammation, endothelial-to-mesenchymal transition, osteogenic reprogramming, valve calcification, and cardiac remodeling. Importantly, pharmacological inhibition of DPP4 with sitagliptin attenuated these pathological features in experimental models.
These findings are particularly relevant because DPP4 inhibitors are already widely used in clinical practice, supporting future retrospective and prospective studies to evaluate their potential impact on CAVD progression and clinical outcomes.
Publication commented by:
Dr María Delgado-Marín & Dr Marta Saura
CARDIOVASCULAR DISEASES. IRYCIS